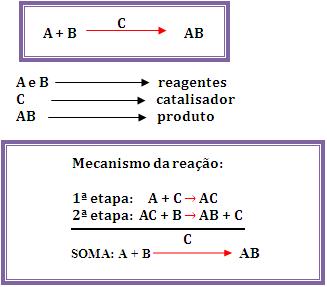
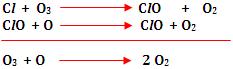
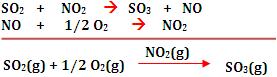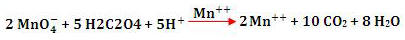É um fato observado que, através do Universo, a energia tende a ser dissipada de tal modo que a energia total utilizável se torna cada vez mais desordenada e mais difícil de captar e utilizar.
Quando conduzimos uma carro a energia armazenada na gasolina é convertida em calor por combustão e, depois, em energia mecânica, no motor. A energia mecânica, ordenada, assim produzida, dá origem ao movimento controlado e ordenado do carro. Mas parte dessa energia foi irrevogavelmente dissipada sob a forma de calor, na estrada, como resultado do atrito dos pneus, no aquecimento do ar por meio da exaustão de gases e para vencer a resistência do vento. Perdemos essa energia para sempre.

A extensão do estado de desordem em que esta energia se encontra é medida por uma quantidade conhecida por entropia. Quanto maior é o estado de desorganização, tanto maior é a entropia, quanto menos extensa for a desorganização, menor é a entropia. De fato, como estabelece a termodinâmica, à temperatura de zero absoluto quando todas as vibrações atômicas e movimento param, a entropia é nula, porque não há movimento desordenado.
Outro exemplo: Suponha que temos água vermelha, com tinta, e água branca, sem tinta, em um tanque, com uma separação. Removendo delicadamente a separação, a água começa dividida, vermelha de um lado e branca do outro. Com o passar do tempo, a água vai gradativamente misturando-se, e no final temos água avermelhada, com a tinta uniformemente distribuída. Agora, mesmo que observemos a mistura por um longo período de tempo, ela não vai separar-se espontaneamente.
A energia total do Universo tende a se tornar cada vez mais desordenada e, por consequência, podemos afirmar que a entropia do Universo cresce continuamente.

Fonte: br.geocities.com
ENTROPIA
Estado de Equilíbrio Termodinâmico
Um certo corpo desliza sobre uma superfície plana. Devido ao atrito, a energia cinética do corpo diminui e, simultaneamente, a sua energia interna e a da superfície aumentam. A energia interna do corpo (e da superfície) é a soma das energias cinéticas do movimento desordenado (microscópico) de cada átomo que constitui esse corpo (ou essa superfície) e das energias potenciais devido às interações mútuas entre esses mesmos átomos. Cada átomo pode ficar, em princípio, com qualquer parcela da energia cinética inicial do corpo, desde que a energia total do sistema corpo + superfície permaneça constante. E como é imensamente grande o número de átomos de qualquer corpo macroscópico, a parte da energia cinética inicial do corpo em questão que fica com ele na forma de energia interna pode ser distribuída de um imenso número de modos entre seus átomos. Assim, qualquer estado do corpo em questão pode ser realizado de um número de modos microscópicos imensamente maior do que qualquer estado anterior, no qual os movimentos dos átomos são ordenados em maior grau, já que o corpo se desloca como um todo com velocidade decrescente.
A passagem espontânea, para o corpo em questão, de um estado a outro significa, com uma probabilidade extremamente grande, a passagem de um estado que pode ser realizado de um certo número de modos microscópicos a um estado que pode ser realizado de um número muito maior de modos microscópicos. E o estado mais provável é aquele que pode se realizar do maior número possível desses modos. Este é o estado de equilíbrio termodinâmico de tal corpo.
Irreversibilidade Termodinâmica
Segundo as leis de Newton, para cada movimento possível de um corpo, existe, sempre, outro movimento, inverso. Em outras palavras, se um corpo pode se deslocar no espaço segundo um movimento direto, digamos, do ponto A ao ponto B, ele também pode se deslocar do ponto B ao ponto A, passando pelos mesmos pontos do espaço e tendo, em cada ponto, a mesma velocidade que no movimento direto, só que em sentido oposto. Como exemplo, seja um projétil lançado do ponto A da superfície da Terra com uma velocidade de módulo v e fazendo um ângulo q com a horizontal. Se a resistência do ar pode ser desprezada, esse projétil atinge o ponto B da superfície da Terra com uma velocidade de módulo v e fazendo um ângulo p - q com a horizontal. Então, se o mesmo projétil é lançado do ponto B com uma velocidade de módulo v e fazendo um ângulo p - q com a horizontal, ele percorre a mesm a trajetória, só que em sentido inverso, e cai no ponto A com uma velocidade de módulo v e fazendo um ângulo q com a horizontal.
Os fenômenos termodinâmicos, por outro lado, são irreversíveis. Se um sistema termodinâmico isolado é abandonado em um certo estado de não equilíbrio, o seu novo estado, no instante seguinte, será, muito provavelmente, um estado que pode ser realizado por um número maior de modos microscópicos e a tendência do sistema é a de se aproximar cada vez mais do estado de equilíbrio termodinâmico. E, uma vez atingido tal estado de equilíbrio, é muito pouco provável que o corpo saia desse estado. A irreversibilidade dos processos termodinâmicos tem um caráter probabilístico, isto é, a passagem espontânea do sistema de um estado de equilíbrio a um estado de não equilíbrio, estritamente falando, não é impossível, mas é tanto mais improvável quanto maior o número de partículas que constituem o sistema.
Para discutir essa última afirmação, seja um recipiente dividido em duas partes iguais contendo numa delas uma certa quantidade de gás e na outra, vácuo. Ao se abrir uma passagem na parede de separação, a metade antes evacuada é preenchida com gás. O processo inverso, a saída espontânea de todo gás de uma das metades do recipiente para a outra, nunca se realiza se o número de partículas é grande. Como cada molécula do gás permanece, em média, o mesmo tempo em cada uma das duas metades do recipiente, a probabilidade de encontrar qualquer molécula numa delas é 1/2. Se o gás em questão pode ser considerado ideal, cada molécula se move independentemente das demais e a probabilidade de encontrar duas moléculas dadas na mesma metade do recipiente (1/2)2, a probabilidade de encontrar três moléculas dadas na mesma metade do recipiente é (1/2)3, etc., de modo que a probabilidade de encontrar todas as N moléculas do gás na mesma metade do recipiente é (1/2)N. Assim, se existem 100 moléculas, por exemplo, a probabilidade de encontrá-las todas na mesma metade do recipiente é (1/2)100 7,9 x 10-31. E se fosse possível medir a posição dessas 100 moléculas uma vez a cada segundo, existe uma chance a cada (1/2)-100 segundos de encontrar todas elas numa metade do recipiente, ou seja, uma chance a cada 1,27 x 1030 segundos ou uma chance a cada 4,03 x 1022 anos. Qualquer sistema macroscópico está constituído de um número de partículas da ordem do número de Avogadro, cerca de 6 x 1023 moléculas. Portanto, a afirmativa de que não é impossível a passagem espontânea do sistema de um estado de equilíbrio a um estado de não equilíbrio, embora verdadeira, é apenas formal.
Entropia
O número de modos microscópicos com que um estado termodinâmico de um sistema pode ser realizado, e que representaremos por W, define a tendência desse sistema passar a outros estados termodinâmicos. O sistema, abandonado a si mesmo, tende a passar de um estado a outro onde W é maior.
A grandeza definida por:
S = k ln W
onde k 1,38 x 10-23 JK-1 é a constante de Boltzmann, é o que se chama de entropia do sistema. O número de modos microscópicos com que se pode realizar o estado de um sistema composto de dois subsistemas, por exemplo, é W = W1 W2, onde W1 e W2 são os números de modos microscópicos com que se pode realizar os estados dos dois subsistemas em questão. Então:
S = k ln W = k ln [ W1 W2 ] = k ln W1 + k ln W2 = S1 + S2
Assim, a entropia de um sistema composto é a soma das entropias de suas partes. Para que esta propriedade termodinâmica da entropia seja realizada é que entra o logaritmo na definição de entropia. Considerando uma variação infinitesimal no estado de um sistema, a correspondente variação infinitesimal de entropia (DS) se relaciona à quantidade infinitesimal de energia absorvida ou perdida na forma de calor (Q) e à temperatura absoluta (T) pela relação termodinâmica:
DS Q / T
onde a igualdade vale se a variação no estado do sistema é reversível e a desigualdade, se a variação é irreversível.
Fisicamente, essa relação se justifica do seguinte modo. Como a energia absorvida por um sistema na forma de calor aparece como energia interna desse sistema, ou seja, aparece nos movimentos microscópicos associados aos átomos e/ou moléculas desse sistema, e isso aumenta o número de modos microscópicos com que o novo estado do sistema pode ser realizado, a variação na entropia do sistema deve ser proporcional à quantidade de energia absorvida na forma de calor: DS Q. Além disso, dada uma certa quantidade de energia Q absorvida na forma de calor, a variação da entropia do sistema deve ser tanto menor quanto maior a energia interna do sistema, e como a energia interna do corpo é medida pela sua temperatura absoluta, a variação na entropia do sistema deve ser inversamente proporcional a essa temperatura: DS T-1.
Segunda Lei da Termodinâmica
A segunda lei da Termodinâmica determina o sentido da evolução dos processos termodinâmicos. Essa lei pode ser formulada em termos da entropia. A entropia de um sistema isolado nunca decresce: não se altera nos processos reversíveis e aumenta nos processos irreversíveis que ocorrem dentro do sistema. O estado de equilíbrio termodinâmico do sistema é o estado de máxima entropia.
O aumento da entropia em processos irreversíveis é muito importante para dar sentido ao próprio conceito de entropia. A energia e a entropia de um sistema isolado não variam se o sistema evolui reversivelmente. Por definição, em qualquer estágio de um processo reversível, o sistema deve estar em um estado de equilíbrio termodinâmico. E como leva um certo tempo para que o sistema, uma vez perturbado, atinja um novo estado de equilíbrio termodinâmico, um processo só pode ser completamente reversível se se desenvolver muito lentamente. Isso, obviamente, nunca acontece. Por outro lado, a energia se conserva e a entropia sempre aumenta nos processos irreversíveis que ocorrem num sistema isolado. A propriedade de conservação da energia, sendo inerente a um sistema isolado, quaisquer que sejam os processos, reversíveis ou não, pelos quais passa o sistema, mostra que a energia não pode indicar o sentido da evolução de tais processos. Mas, o aumento da entropia nos processos irreversíveis, aumento esse também inerente a um sistema isolado, mostra que a entropia pode indicar, sim, o sentido da evolução de tais processos: o estado inicial pode ser diferenciado do estado final porque este tem, necessariamente, maior entropia.
Fonte: br.geocities.com
ENTROPIA
Entropia é a medida da "quantidade de desordem" de um sistema. Muita desordem implica uma entropia elevada ao passo que a ordem implica uma baixa entropia. Não é difícil compreender o motivo desta associação já que a entropia de uma sustância no estado gasoso é superior à entropia da mesma substância no estado líquido, que é maior que no estado sólido... E as moléculas estão mais ordenadas no estado sólido e mais dispersas e caóticas no estado gasoso, sendo o estado líquido um estado intermédio.
Do mesmo modo, numa divisão onde haja objectos espalhados desordenadamente pelo chão, a entropia é superior à da mesma divisão onde esses objectos estão arrumadinhos em locais devidamente adequados. Assim se percebe a associação entropia/desordem... Uma boa desculpa para quem se desleixa na arrumação do seu quarto! De acordo com o Segundo Princípio da Termodinâmica, a diminuição da entropia num espaço, equivale ao aumento da mesma na pessoa que gasta energia a arrumar!!!
Segundo Princípio da Termodinâmica
O Segundo Princípio da Termodinâmica diz precisamente que um sistema isolado tende a evoluir no sentido de aumentar a entropia. Aqui está a explicação para o facto de as coisas acontecerem assim e não ao contrário... É que a entropia do universo aumenta sempre e os acontecimentos inversos implicariam a diminuição de entropia!
Mas que estou eu a dizer?
Se a entropia nunca diminui como é possível a formação de gelo? A entropia da água diminui quando ela passa ao estado sólido!!! Será esta uma incompatibilidade da teoria?
A chave aqui é a palavra "universo". A entropia pode diminuir em algumas coisas se aumentar noutras. Assim se explica a formação de gelo! Se colocares água a 20ºC no congelador a –5ºC, o calor flui da água para o congelador, aumentando a entropia do sistema e diminuindo a entropia da água. Na verdade, a entropia total do universo aumenta.
Se a formação do gelo fosse um processo natural, o Segundo Princípio da Termodinâmica seria violado. Mas isso não acontece... O teu congelador não funciona se não lhe forneceres energia para que o motor funcione, acabando por produzir calor que se dispersa pela tua cozinha, aumentando a entropia total do universo...
O Princípio está salvo!
Quando usas uma pequena bomba manual para encher o pneu da tua bicicleta ou a tua bola de volei, estás a deslocar o ar de uma região onde está relativamente "espalhado" para um local onde é muito mais denso. Isto representa uma diminuição de entropia, mas, tal como no frigorífico, este processo não acontece sozinho: implica um trabalho da tua parte, trabalho esse que exige energia. O produto total desse processo é o calor que acaba por aumentar a entropia do universo.
O Segundo Princípio da Termodinâmica não defende que a entropia não possa diminuir num determinado local, ela tem é de aumentar noutro lado! Fascinante, não achas?
Entropia vs. Energia
Um dos factos mais curiosos do aumento da entropia do universo, atrás referido, é a consequente "degradação" da energia!
Como vimos anteriormente, sempre que ocorre uma transformação irreversível dá-se um aumento da entropia do universo, mas por outro lado perdemos a oportunidade de obter energia sob forma utilizável.... ou seja, a energia que foi convertida em trabalho para que o processo se desenrolasse, embora não tenha sido "destruída", encontra-se "degradada", não podendo mais ser utilizada para obtermos trabalho útil! Daí que quando falamos de poupança de energia estejamos a falar em poupança de energia utilizável, porque que a energia se conserva já sabemos há bastante tempo!!
Confuso?!!!??? Vamos ver um exemplo para aclarar ideias!
O "Café com Leite" (o meu exemplo favorito) vai ajudar-nos...
Se adicionarmos o café quente ao leite frio teremos um sistema irreversível que irá evoluir de forma a obter uma temperatura uniforme. Hummm... temos um "café com leite" óptimo... e não só:
Ocorreu um aumento da entropia do universo!
No entanto, existe um "mas"..... e se tivéssemos utilizado as duas fracções (quente e fria) da nossa bebida para obter trabalho a partir de uma qualquer máquina térmica? Pois, afinal entropia é inversamente proporcional à energia disponível!
Sadi Carnot mostrou acreditar piamente no calórico, um fluido que passaria dos corpos "mais quentes" para os "mais frios". Carnot observou também que as máquinas térmicas eram cíclicas, voltando repetidamente ao estado inicial, e que para funcionarem precisavam não só de uma fonte quente de onde extraíam calor (caldeira), como de uma fonte fria para onde o enviavam (condensador). Assim, de acordo com o Segundo Princípio da Termodinâmica, não existe nenhuma máquina térmica que se limite a produzir trabalho.
Mas voltemos à questão do café com leite: o que aconteceu realmente na bebida?
Após a bebida estar preparada a sua temperatura tornou-se uniforme, perdemos a fonte quente (café) e a fonte fria (leite), e perdemos assim a hipótese de transformar calor em trabalho. No sistema a energia total mantêm-se de acordo com a Lei da Conservação da Energia (Primeiro Princípio da Termodinâmica) mas a entropia aumenta dado ser um sistema irreversível!!!!!!!
Voltemos agora ao universo!
Pelo que observamos do princípio do aumento da entropia, concluímos que o nosso universo, em virtude dos processos naturais, tende para um estado de desordem maior e uniformidade geral!
E que significa isso?
À medida que ocorrem esses processos a energia disponível para efectuar trabalho útil ira diminuir! Todos os processos físicos, químicos e biológicos cessarão, atingindo uma situação limite a que geralmente se dá o nome de "Morte Térmica" do universo.
Estas são as conclusões que podemos prever, a partir dos nossos conhecimentos actuais acerca da termodinâmica... no entanto não te assustes, pois uma situação extrema a ocorrer estará num futuro bem distante!!!
Fonte: www.ajc.pt